


Business Inquiry
Global:
Email:marketing@medicilon.com
+1(781)535-1428(U.S.)
0044 7790 816 954 (Europe)
China:
Email: marketing@medicilon.com.cn
Tel: +86 (21) 5859-1500



Sickle-cell disease falls into an intriguing scientific paradigm—it is very well studied from multiple scientific angles and most of the molecular mechanisms associated with the disease have been elucidated. However, treatments for the disease have been lacking, as there are very few ways to combat the genetic disorder, without rectifying the DNA mutation that causes the sickle-cell trait.
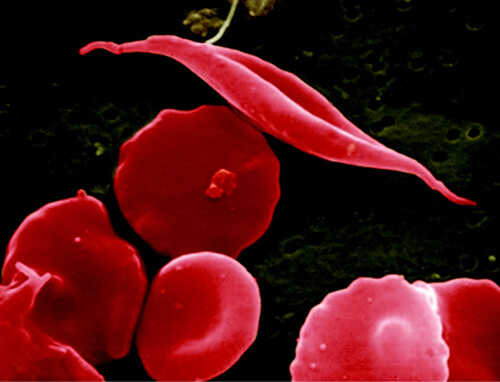
Yet now, researchers from Dana-Farber/Boston Children’s Cancer and Blood Disorders Center have reported on a precision-engineered gene therapy virus that is inserted into blood stem cells and then transplanted, which markedly reduced sickle-induced red-cell damage in mice with sickle cell disease. This work stems off a decades-old discovery about sickle-cell disease, which found that the illness is milder in people whose red blood cells carry a fetal form of hemoglobin.
Sickle cell disease is caused by a mutation in hemoglobin, the oxygen-carrying protein in red blood cells, which causes the usually pliant cells to stiffen and assume a curved or “sickled” shape. These sickled cells live less long than those with healthy hemoglobin, leading to anemia. They also bind together, resulting in a blockage of blood flow that can cause severe pain and organ damage throughout the body.
Researchers have known that fetal hemoglobin typically tails off after birth, but in 2008 investigators found that suppressing a gene called BCL11A—which acts as an “off” switch—could restart fetal hemoglobin production. In 2011, using this approach, the same group corrected sickle cell disease in mice, replacing much of the defective beta (“adult”) hemoglobin that causes sickling with healthy fetal hemoglobin.
In order to turn this discovery into a clinical therapy, scientists need to overcome a major hurdle: BCL11A has a major role in blood stem cells. When it is silenced in mice with the sickle cell mutation, blood stem cells cannot engraft long-term in the animals’ bone marrow and eventually become depleted. This failure to engraft could render gene therapy ineffective and also cause serious problems with blood development in general.
To circumvent this problem the Dana-Farber/Boston Children’s team performed some clever genetic engineering, creating a gene therapy virus that silences BCL11A selectively—only in precursors of red blood cells. Blood stem cells treated with this gene therapy, transplanted into the mice, engrafted successfully and reduced signs of sickle cell disease (hemolytic anemia and increased numbers of reticulocytes).
Interestingly, the researchers also found that in red blood cells from the mice and four patients with sickle cell disease, fetal hemoglobin crowded out the sickling beta hemoglobin, making up at least 80% of the total hemoglobin in the cell—more than enough to avoid clinical sickle cell disease.
“The tendency for a red blood cell to sickle is proportional to how much non-sickling versus sickling hemoglobin it has,” explained senior study investigator David A. Williams, M.D., chief of the Division of Hematology/Oncology at Boston Children’s Hospital. “BCL11A represses fetal hemoglobin, which does not lead to sickling, and also activates beta hemoglobin, which is affected by the sickle-cell mutation. So when you knock BCL11A down, you simultaneously increase fetal hemoglobin and repress sickling hemoglobin, which is why we think this is the best approach to gene therapy in sickle cell disease.”
Selectively knocking down BCL11A involved several layers of engineering. As the core of their gene therapy vector, the researchers used a snippet of genetic code called a short hairpin RNA (shRNA) that inactivates the BCL11A gene. To get it into cells, they embedded the shRNA in another bit of code, a microRNA, that cells recognize and process. To make this assembly work in the right place at the right time, they hooked it to a promoter of beta hemoglobin expression, together with regulatory elements active only in precursors of red blood cells. Finally, they inserted the whole package into a lentivirus engineered for safe use in humans.
“We found that using optimized shRNAs embedded within an miRNA (shRNAmiR) architecture to achieve ubiquitous knockdown of BCL11A profoundly impaired long-term engraftment of both human and mouse hematopoietic stem cells (HSCs) despite a reduction in nonspecific cellular toxicities,” the authors wrote. “BCL11A knockdown was associated with a substantial increase in S/G2-phase human HSCs after engraftment into immunodeficient (NSG) mice, a phenotype that is associated with HSC exhaustion.”
The findings from this study were published recently in the Journal of Clinical Investigation through an article entitled “Lineage-specific BCL11A knockdown circumvents toxicities and reverses sickle phenotype.”
The researchers believe the BCL11A-based approach to gene therapy for sickle cell disease will substantially increase the ratio of non-sickling versus sickling hemoglobin. Moreover, they suggest that this approach may also be beneficial in beta-thalassemia, another blood disorder involving defective beta hemoglobin. Currently, the scientists are taking the final steps toward FDA clearance for a clinical gene therapy trial in sickle cell disease that is expected to begin in early 2017.
 Share:
Share:
 Relevant
news
Relevant
news







 marketing@medicilon.com
marketing@medicilon.com


